
Конечностно-поясная (поясно-конечностная) мышечная дистрофия R9 / 2i – это аутосомно-рецессивная мышечная дистрофия, вызванная мутациями в гене FKRP (fukutin-related protein – фукутин-связанный белок).
КПМД 2i может проявиться в разном возрасте – и в раннем подростковом, и в пожилом – после 70-ти лет.
В 2017 году была разработана новая классификация конечностно-поясных мышечных дистрофий (см. подробнее о новой классификации). По этой классификации КПМД 2i присвоено обозначение R9, что означает рецессивное наследование (R) и 9-й номер по очередности открытия подтипов КПМД.
Внимание! Если у вас во врачебных заключениях стоит миопатия/миодистрофия (и т.п.) Эрба-Рота, значит, диагноз вам не поставлен. Добивайтесь точной постановки диагноза с генетическим подтверждением! Это поможет подобрать правильную терапию и предотвратить или уменьшить последствия заболевания.
Содержание страницы
- Альтернативные названия
- Повреждённый ген и белок
- Частота заболевания
- Пациентское сообщество
- Фонды и реестр (регистр) пациентов
- Исследования
- Диагностика, симптомы и течение заболевания
Альтернативные названия
В тексте использую строчную букву i, чтобы зрительно было легче воспринимать обозначение этой формы, т.к. прописная I слишком похожа на строчную L.
- конечностно-поясная мышечная дистрофия R9
- поясно-конечностная мышечная дистрофия R9 / 2i
- КПМД (ПКМД) R9 / 2i
- КПМД, связанная с дефицитом FKRP
- аутосомно-рецессивная КПМД 2i / R9
- limb-girdle muscular dystrophy 2i (LGMD2i)
- limb-girdle muscular dystrophy R9 (LGMDR9) FKRP-related
- LGMD due to FKRP deficiency
- Autosomal recessive limb-girdle muscular dystrophy type 2i
- muscular dystrophy-dystroglycanopathy (limb-girdle) type C, 5; MDDGC5
- дистрогликанопатия (dystroglycanopathy). Имейте в виду, что к дистрогликанопатиям относятся несколько заболеваний (в справочнике OMIM введите dystroglycanopathy в поиске)
Повреждённый ген и белок
Ген, поломки в котором приводят к КПМД R9/2i: FKRP (страница гена FKRP в справочнике OMIM).
Белок, который формируется из гена: fukutin-related protein (FKRP), фукутин-связанный белок.
Следует различать фукутин (FKTN) и фукутин-связанный белок (FKRP).
От мутации к мышечной слабости
В 2016 году было сделано открытие, что фукутин-связанный белок модицифирует белок, находящийся в мембране наших мышц. Было сделано предположение, что модификации мембранного белка, сделанные фукутин-связанным белком, критически важны для поддержания целостности мышц во время их сокращения и расслабления в процессе, когда человек двигается, встаёт, дышит.
У людей с КПМД 2i мутация гена FKRP влияет на функциональность кодируемого им белка. Если фукутин-связанный белок сформирован неправильно, то это значительно уменьшает его эффективность. В результате, фукутин-связанный белок не может нормально модифицировать целевой белок (с которым FKRP должен взаимодействовать). Предполагается, что это и есть главная причина, из-за которой мышцы при КПМД 2i становятся хрупкими и разрушаются с гораздо бо́льшей скоростью по сравнению с мышцами людей, у которых нет мутации в гене FKRP [1].
Хрупкие мышцы
В разделе о мышцах на сайте LGMD2iFund разъясняется: все миофибриллы покрыты базальной мембраной. Для того чтобы базальная мембрана укрепляла волокна и сохраняла их от повреждения в процессе движения, необходимо, чтобы она была химически связана с волокном. Один из основных белков миофибрилл, связанный с базальной мембраной, называется альфа-дистрогликан (alpha-dystroglycan (aDG)). Альфа-дистрогликан должен быть модифицирован фукутин-связанным белком, чтобы нормально функционировать. Предполагается, что в результате неправильной работы FKRP не происходит модифицирования альфа-дистрогликана, который, в свою очередь, не может взаимодействовать с базальной мембраной, тем самым увеличивая уязвимость миофибрилл в процессе циклов мышечного сокращения и расслабления.
Потеря целостности мембраны вследствие повреждений из-за мышечных сокращений в конечном итоге приводит к разрушению мышц [1].
Ниже приведены иллюстрации последствий повреждения здоровой мышцы и мышцы с дистрофией (источник DOI 10.1186/2044-5040-1-21).
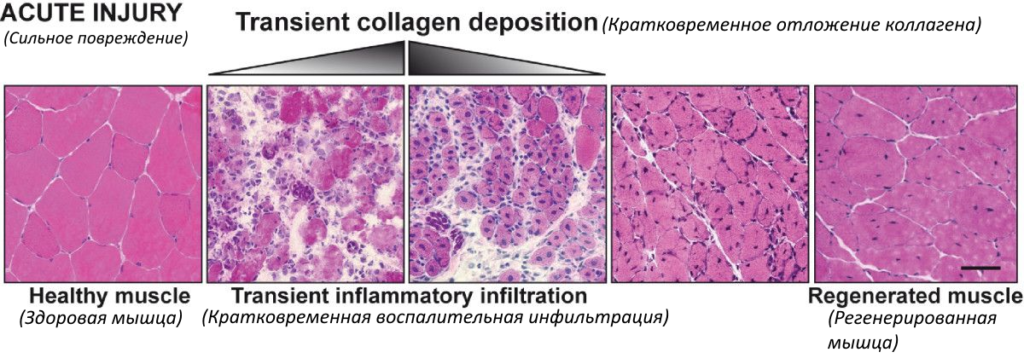
При сильном повреждении в здоровой мышце происходит кратковременное отложение коллагена (на рисунке белым цветом) и быстрое и контролируемое воспаление, которое удаляет мёртвые и повреждённые мышечные волокна и способствует замене повреждённой мышцы [1].
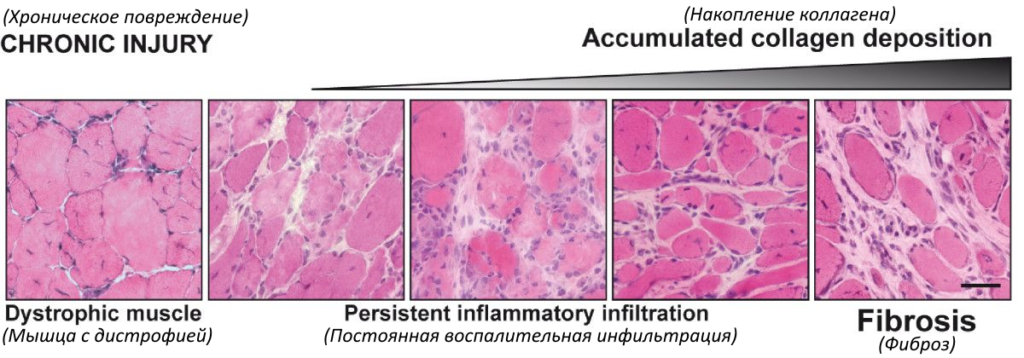
В состоянии хронического повреждения, как это происходит при мышечных дистрофиях, хроническое воспаление приводит к чрезмерному накоплению компонентов внеклеточного матрикса, что препятствует миогенному восстановлению и приводит к замещению мышечной ткани на фиброзную / рубцовую ткань [1].
Частота заболевания
Общая распространенность конечностно-поясной мышечной дистрофии R9/2i по данным Orphanet: 1-9 / 100 000.
Пациентское сообщество
Есть русскоязычная пациентская группа по конечностно-поясной мышечной дистрофии R9 / 2i.
Если у вас генетически подтверждённая КПМД R9/2i, для вступления в пациентскую группу свяжитесь с администратором группы – Ольгой Киряковой (страница Вк).
Фонды и реестр (регистр) пациентов
Людям с заболеваниями, вызванными мутациями в гене FKRP, полезно будет зарегистрироваться в международном регистре пациентов с мутациями в гене FKRP.
Организация, ведущая этот регистр, регулярно оповещает участников о новостях, связанных с поиском лечения заболеваний, вызванных мутациями в этом гене.
Почему нужно регистрироваться в реестре пациентов?
Для редких заболеваний такие регистры особенно важны, т.к. разработчикам препаратов важно знать количество пациентов, страны, где это заболевание чаще встречается (что в дальнейшем повлияет на решение о проведении клинических испытаний), виды мутаций (для генетических болезней) и другие особенности.
Регистры помогают привлечь внимание к проблемам, увеличить финансирование (как с помощью благотворительности, так и с помощью научных грантов), поднять уровень осведомлённости о заболевании.
Исследования
На отдельной странице я собираю информацию по препаратам, разрабатываемым для лечения конечностно-поясных мышечных дистрофий. Нажмите для перехода к разделу о разработках лечения КПМД 2i.
Ниже привожу более подробную информацию о конечностно-поясной мышечной дистрофии R9 / 2i типа.
Диагностика, симптомы и течение заболевания
В 2013 году в журнале «Нервно-мышечные болезни» было опубликовано исследование «Сравнительный анализ особенностей фенотипов поясно-конечностных мышечных дистрофий 2А и 2I типов». Авторы исследования отмечают, что, несмотря, на высокую степень сходства фенотипов больных с КПМД, существуют особенности, которые помогут при постановке верного диагноза. На странице о КПМД 2A (см. раздел “Диагностика”) я приводила выдержки из этой статьи, поэтому дублировать тут их не буду.
Симптомы КПМД 2i
Ниже приведён список симптомов. Учитывайте, что симптомы могут различаться у пациентов. Информация взята из базы данных Human Phenotype Ontology (HPO). В этой базе данных собирается информация о симптомах болезней, описанных в разных медицинских источниках. HPO обновляется регулярно. По идентификатору ID HPO вы можете найти больше информации по каждому симптому [2]. Также по этому идентификатору можно посмотреть названия (и синонимы) на английском языке.
Обращение к врачу, который столкнулся с редким пациентом и зашёл на эту страничку в поисках информации, – спасибо за небезраличие и возьмите на заметку базу данных HPO и сайт GARD 😉
Симптомы в 80-99% случаев:
- Повышенный уровень креатинкиназы (креатинфосфокиназа (КФК)) – ID HPO 0003236
- Мышечная дистрофия – ID HPO 0003560
- Проксимальная мышечная слабость (cлабость в верхних частях рук и ног) – ID HPO 0003701
- Снижение уровня альфа-дистрогликана в мышечных волокнах – ID HPO 0030099
Симптомы в 30-79% случаев:
- Аномалии в ахиллесовом сухожилии – ID HPO 0005109
- Гипертрофия икроножных мышц (увеличенный размер икроножных мышц) – IP HPO 0008981
- Генерализованная гипотония (пониженный или низкий мышечный тонус) – ID HPO 0001290
- Слабость мышц тазового пояса – ID HPO 0003749
- Слабость мышц плечевого пояса (слабые плечевые мышцы) – ID HPO 0003547
- Раскачивающаяся походка (среди русскоязычных врачей распространено – “утиная* походка”) – ID HPO 0002515
Симптомы в 5-29% случаев:
- Сложно подниматься по лестнице – ID HPO 0003551
- Сложно бегать – ID HPO 0009046
- Дилатационная кардиомиопатия (растянутая и истончённая сердечная мышца) – ID HPO 0001644
- Частые падения – ID HPO 0002359
- Пониженная моторика – ID HPO 0001270
- Снижение уровня мерозина в мышечных волокнах – ID HPO 0030092
- Сколиоз (искривление позвоночника) – ID HPO 0002650
Симптомы в 1-4% случаев:
- Миоглобинурия, вызванная физической нагрузкой – ID HPO 0008305
Процент не указан на HPO:
- Контрактуры (укорочение, жёсткость) ахиллесова сухожилия – ID HPO 0001771
- Аутосомно-рецессивное наследование – ID HPO 0000007
- Врождённая мышечная дистрофия – ID HPO 0003741
- Сложности при ходьбе – ID HPO 0002355
- Гиперлордоз (искривление в поясничном отделе позвоночника) – ID HPO 0003307
- Кифоз (искривление в грудном или пояснично-грудном отделе позвоночника) – ID HPO 0002808
- Дисфункция левого желудочка – ID HPO 0005162
- Макроглоссия (аномально большой язык) – ID HPO 0000158
- Мышечные спазмы – ID HPO 0003394
- Миалгия (мышечные боли) – ID HPO 0003326
- Ночная гиповентиляция – ID HPO 0002877
- Дыхательная недостаточность – ID HPO 0002111
- Гипертрофия (увеличенный размер) бедренных мышц – ID HPO 0003733
- Ходьба на мысочках – ID HPO 0040083
- Вариации в проявлении симптомов – ID HPO 0003828
- Срастание позвонков (Vertebral/spinal fusion) – ID HPO 0002948
До сих пор не ясно, почему некоторые группы мышц более подвержены разрушению при неправильной работе фукутин-связанного белка по сравнению с другими группами и почему в одной и той же мышечной группе определённые мышцы разрушаются быстрее и сильнее, чем другие.
Мышечная слабость при КПМД 2i сначала влияет на способность подниматься с пола и подниматься по лестнице, а также на способность удерживать баланс тела.
Физиотерапия*, упражнения** и некоторые методы альтернативной терапии считаются способом замедлить прогрессирование КПМД 2i [1]
*Термин “Физиотерапия” имеет различные значения в западной медицине и на постсоветском пространстве. Например, см. Википедию о физиотерапии. Поскольку источник статьи – зарубежный, то информацию о физиотерапии ищите в контексте западной медицины.
**Внимание! Упражнения при мышечных дистрофиях должны быть щадящими, и ни в коем случае нельзя перенапрягать мышцы!
Сердечные и дыхательные осложнения
Вдобавок к мышцам конечностей, при мышечной дистрофии 2i также страдают мышцы диафрагмы и сердца. Степень и скорость повреждения сердечной мышцы и мышц диафрагмы не предсказуемы. Однако, поскольку повреждение этих органов является распространённой чертой КПМД 2i, каждый пациент с этим заболеванием должен регулярно наблюдаться у кардиолога и пульмонолога.
Из-за того что дыхательная и сердечная недостаточность могут развиться до момента потери способности передвигаться, целесообразно проводить сердечную и дыхательную оценку во время диагностики. Данная первичная оценка этих функций даст отправную точку для сравнения ваших показателей сердечной и дыхательной способности в будущем.
Люди могут не осознавать значительную потерю сердечной и дыхательной функций, пока они не станут очень заметными:
- дыхательная недостаточность приводит к ночной гиповентиляции лёгких – состоянию, при котором (кроме других симптомов) возникает общая вялость и сонливость в течение дня, а также усталость и головные боли по утрам;
- некоторыми признаками развивающейся кардиомиопатии являются нехватка воздуха, усталость, сильное сердцебиение, головокружение и боль в груди [1].
Источники
- About LGMD2I на сайте LGMD2IFund
- Limb-girdle muscular dystrophy type 2I на сайте Genetic and Rare Diseases Information Center (GARD)
- Страница о гене FKRP и о LGMDR9 в справочнике OMIM
- Страница о LGMDR9 в справочнике Orphanet.
Внимание
Для сайта я подбираю материалы из различных источников, которые стараюсь максимально проверить на достоверность и научную значимость. Ссылки на источники размещаю в скобках после абзацев или в конце страницы/записи. Однако приведенную информацию нельзя рассматривать как абсолютно достоверный медицинский источник. Обязательно консультируйтесь со специалистами. Англо/франкоязычные тексты перевожу я сама. Я не врач. Если Вы нашли ошибку, неточность или хотите дополнить информацию, пожалуйста, напишите мне на электронную почту lgmd.ru@yandex.ru.
При использовании материалов сайта lgmd.ru обязательно указывайте активную ссылку на источник – сайт lgmd.ru.
Добавить комментарий